
Il Sindrome di Waardenburg (SW) è una patologia di origine genetica classificata come un tipo di neuropatia. Le sue caratteristiche cliniche sono definite dalla presenza di sordità o perdita dell'udito, pigmentazione anormale degli occhi, dei capelli o della pelle e varie alterazioni facciali.
Questa patologia è caratterizzata dalla sua ampia variabilità sintomatica, motivo per cui si distinguono diversi tipi: Tipo I, Tipo II, Tipo III (sindrome di Klein-Waardenburg o psudo Waardenburg) e Tipo IV.

A livello eziologico, la sindrome di Waardenburg ha un pattern ereditario autosomico dominante. Di solito è associato a mutazioni specifiche nei geni EDN3, EDNRB, PAX3, SOX10, SNAI2 e MIT..
La diagnosi viene effettuata sulla base di vari criteri clinici maggiori e minori. Tuttavia, è necessario eseguire vari test di laboratorio complementari. Non esiste una cura o un trattamento specifico per la sindrome di Waardenburg..
L'intervento con questa patologia si concentra solitamente sul trattamento dei disturbi dell'udito (procedure chirurgiche, impianti cocleari, ecc.), Logopedia e riabilitazione neuropsicologica, oltre che psicologica.
Indice articolo
Questa sindrome fu inizialmente descritta dal genetista e oftalmologo olandese Petrus Johannes Waardenburg nel 1848. Nel suo rapporto clinico fece riferimento alle principali caratteristiche cliniche:
Successive analisi hanno identificato un'ampia variabilità clinica nella sindrome di Waardenbur. Inoltre, Mckusick ha associato questa sindrome ad altri decorsi clinici simili, come la malattia di Hirschsprung..
Allo stato attuale, è considerata una patologia rara, che si manifesta con un grado variabile di ipoacusia che può causare alterazioni significative nell'apprendimento e successivo sviluppo della persona interessata.
La prognosi per la sindrome di Waardenburg è favorevole, sebbene possa essere associata a morbilità e mortalità significative legate a complicazioni mediche, soprattutto intestinali.
La sindrome di Waardenburg è una malattia genetica congenita i cui segni e sintomi tendono a variare ampiamente tra le persone colpite..
Le caratteristiche più comuni includono anomalie facciali distintive, pigmentazione alterata della pelle, degli occhi o dei capelli e sordità..
Nella letteratura medica, questa sindrome è spesso considerata un tipo di genodermatosi o neuropatia. Il termine genodermatosi si riferisce a un ampio insieme di malattie caratterizzate dalla presenza di anomalie cutanee e alterazioni di origine genetica..
D'altra parte, il termine neuropatia si riferisce a un gruppo di patologie derivate dallo sviluppo di anomalie e processi difettosi durante la migrazione e il differenziamento delle cellule della cresta neurale durante la gestazione..
La cresta neurale è una struttura embrionale costituita da un ampio insieme di cellule indifferenziate il cui sviluppo porterà alla formazione della struttura cranio-facciale e delle cellule neuronali e gliali che formeranno gran parte del sistema nervoso..
Tra l'ottava e la decima settimana di gestazione, di solito inizia il processo di migrazione delle cellule che compongono la cresta neurale. Quando vari fattori patologici o anomalie genetiche interferiscono in questo processo, possono comparire significative anomalie cognitive e / o fisiche, come la sindrome di Waardenburg..
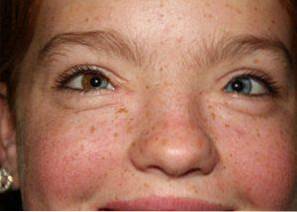
La prevalenza della sindrome di Waardenbur è stimata in 1 caso su 40.000 persone in tutto il mondo. Dalla sua scoperta, sono stati descritti circa 1.400 casi diversi nella letteratura medica e sperimentale..
Sembra colpire allo stesso modo uomini e donne. Non sono state identificate associazioni con regioni geografiche o particolari gruppi etnici e razziali.
La sindrome di Waardenbug rappresenta il 2-5% di tutti i casi diagnosticati di ipoacusia congenita.
Sebbene siano stati identificati vari decorsi clinici, il tipo I e II sono i più comuni. I tipi III e IV sono rari.

La sindrome di Waardenburg è caratterizzata da tre alterazioni fondamentali: alterazioni cranio-facciali, anomalie pigmentarie e sordità:
Un altro dei risultati medici centrali della sindrome di Waardenburg è la perdita della capacità uditiva e dell'acuità. Il più comune è identificare nelle persone colpite un grado variabile di sordità o ipoacusia neurosensoriale.
Il termine ipoacusia neurosensoriale si riferisce a una perdita della capacità uditiva derivante da lesioni interne correlate alle terminazioni nervose che conducono le informazioni uditive dall'orecchio interno ai centri cerebrali..

La sindrome di Waardenburg è classificata in 4 tipi di base in base al decorso clinico e ai sintomi specifici che sono presenti nelle persone affette:
La sindrome di Waardenbuug ha un'origine congenita associata a varie alterazioni genetiche.
L'analisi dei casi ha permesso di individuare queste anomalie nei geni: EDN3, EDNRB, PAX3, SOX10, SNAI2 e MIT.
Questo insieme di geni sembra essere coinvolto nello sviluppo e nella formazione di vari tipi di cellule, comprese quelle responsabili della produzione di melanociti.
I melanociti sono responsabili della generazione di melanina, un pigmento che contribuisce alla colorazione degli occhi, dei capelli o della pelle.
A seconda dei diversi decorsi clinici, possiamo identificare diverse alterazioni genetiche:
Come abbiamo sottolineato nella descrizione iniziale, la diagnosi della sindrome di Waardenbug viene effettuata sulla base di diversi criteri principali e minori:
Per stabilire una diagnosi definitiva, è essenziale identificare la presenza di due criteri principali o almeno uno maggiore e due minori. Inoltre, è necessario utilizzare alcuni test complementari: biopsia, audiometria o test genetici.
Non esiste una cura per la sindrome di Waardenbug, sebbene possano essere utilizzati approcci sintomatici..
Il trattamento dei segni e dei sintomi più comuni di solito richiede l'intervento medico di dermatologi e oftalmologi..
D'altra parte, nel caso del trattamento della sordità neurosensoriale, può essere eseguito un impianto cocleare accompagnato da una logopedia e da un intervento neuropsicologico..



Nessun utente ha ancora commentato questo articolo.