
Il Sindrome di Pallister-Killian, nota anche come tetrasomia 12, è una malattia rara di origine genetica caratterizzata da un ampio spettro di coinvolgimento multiorgano.
Clinicamente, questa patologia è definita da disabilità intellettiva, ritardo psicomotorio, ipotonia muscolare, fenotipo facciale atipico, anomalie pigmentarie della pelle e alopecia. Inoltre, possono comparire anche altri tipi di complicanze mediche legate a malformazioni in diversi sistemi corporei o convulsioni..
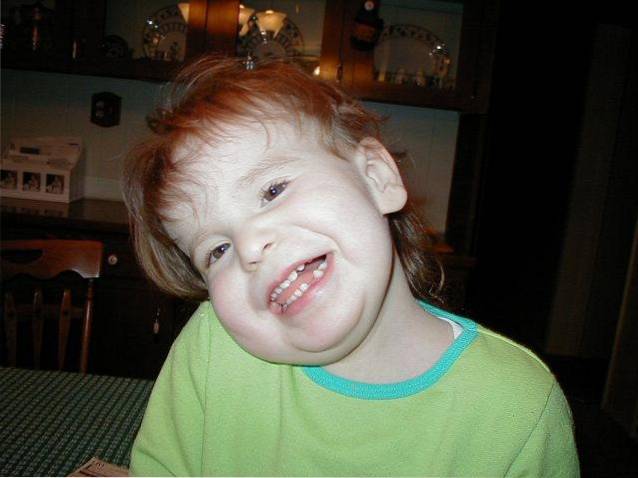
L'origine eziologica di questa malattia è associata a una malattia genetica distribuita a mosaico. In particolare, è dovuto alla presenza di un cromosoma 12 in più in alcune cellule del corpo.
La diagnosi della sindrome di Pallister-Killiam può essere fatta sia prenatale che postnatale. L'obiettivo principale è l'identificazione delle caratteristiche cliniche e l'utilizzo di uno studio genetico di conferma..
Questa sindrome ha un alto tasso di mortalità. Tuttavia, l'approccio medico farmacologico e il trattamento riabilitativo possono fornire importanti benefici per la qualità della vita e lo stato clinico delle persone colpite.
Indice articolo
Questa malattia fu inizialmente descritta da Pallister nel 1977. Nelle prime pubblicazioni, questo ricercatore riportò due casi di pazienti adulti il cui decorso era caratterizzato da vari reperti: convulsioni, ipotonia muscolare, deficit cognitivo, malformazioni muscoloscheletriche e organiche, configurazione ruvida del viso e colore della pelle i cambiamenti.
Parallelamente, Teschler-Nicola e Killiam nel 1981 descrissero questo stesso quadro clinico in una bambina di tre anni.
Pertanto, nelle prime relazioni cliniche, è stato fatto un riferimento generale a una condizione medica caratterizzata dalla combinazione di convulsioni, disabilità intellettiva e un fenotipo fisico caratteristico..
Inoltre già nel 1985 Gilgenkratz riuscì ad identificare nel primo caso durante la fase di gestazione, qualcosa di comune oggi grazie alle moderne tecniche diagnostiche.
La sindrome di Pallister-Killiam è un tipo di malattia del mosaico genetico. In questo caso, l'alterazione cromosomica colpisce solo alcune cellule del corpo. Viene identificato un ampio coinvolgimento di diversi sistemi corporei e organismi.
È caratterizzato principalmente da disabilità intellettiva, ipotonia muscolare, sviluppo di tratti distintivi del viso, alterazione della pigmentazione della pelle o crescita dei capelli, tra le altre alterazioni congenite..
Inoltre, la sindrome di Pallister-Kiliam è una malattia rara di origine congenita che può ricevere un ampio numero di nomi nella letteratura medica:
I dati sulla prevalenza della sindrome di Pallister-Killiam non sono noti con precisione. Non sono state fatte molte diagnosi definitive e la maggior parte di queste non sono state pubblicate nella letteratura medica.
Pertanto, tutti gli autori e le istituzioni definiscono questa sindrome come una patologia genetica rara o poco frequente nella popolazione generale..
Circa 15 anni fa, la sindrome di Pallister-Killiam era stata identificata in quasi 100 casi in tutto il mondo. Attualmente, questa cifra ha superato i 200 interessati.
Le indagini epidemiologiche hanno stimato l'incidenza di questa malattia in circa 5,1 casi per milione di neonati, sebbene autori come Toledo-Bravo de la Laguna e collaboratori la collochino a 1 / 25.000.
Non è stata identificata una maggiore prevalenza associata alle caratteristiche sociodemografiche delle persone colpite. La sindrome di Pallister-Killian può apparire in qualsiasi genere o gruppo tecnico e / o razziale.
Un'ampia varietà di segni e sintomi può essere identificata nel decorso clinico della sindrome di Pallister-Killian. Tutti associati ad anomalie cranio-facciali e / o muscolo-scheletriche e ad alterazioni cognitive.
Lo sviluppo di malformazioni cranio-facciali dalla fase di gestazione alla crescita postnatale e infantile costituisce uno dei segni medici più caratteristici della sindrome di Pallister-Killiam..
I segni e sintomi più comuni includono anomalie nelle diverse strutture craniche e facciali che porteranno ad un aspetto ruvido e atipico:
Nonostante sia meno significativo delle alterazioni facciali, è molto comune osservare diverse anomalie muscolo-scheletriche nei pazienti affetti dalla sindrome di Pallister:
Le anomalie legate alla struttura muscolare e alla mobilità sono un'altra delle caratteristiche cliniche cardinali della sindrome di Pallister-Killian:
L'ipotonia muscolare si riferisce all'identificazione di tono o tensione muscolare anormalmente ridotto. A livello visivo si possono osservare flaccidità e labilità in diversi gruppi muscolari, particolarmente accentuati alle estremità..
Pertanto, la patologia muscolare e scheletrica causerà un significativo ritardo nell'acquisizione di diverse capacità motorie, sia nel periodo neonatale che infantile..
Sebbene i periodi di sviluppo siano variabili tra le persone interessate, il calendario più comune include le seguenti tappe:
Un'altra delle aree fortemente colpite è il sistema nervoso. Nella maggior parte dei casi, i segni ei sintomi sono principalmente correlati a convulsioni e disabilità intellettiva:
Sebbene siano meno frequenti, possono comparire anche altri tipi di complicanze mediche:
L'origine della sindrome di Pallister-Killian è associata a un'anomalia del mosaico genetico sul cromosoma 12. Colpisce solo il materiale genetico di alcune cellule del corpo..
I cromosomi fanno parte del nucleo di tutte le cellule che si trovano nel corpo umano. Sono costituiti da un'ampia varietà di componenti biochimici e contengono le informazioni genetiche di ogni individuo..
Gli esseri umani hanno 46 cromosomi diversi, organizzati in coppie e numerati da 1 a 23. Inoltre, a livello individuale, ogni cromosoma ha un'area o un braccio corto chiamato "p" e uno lungo chiamato "q".
L'anomalia colpisce il cromosoma 12 e provoca la presenza di un cromosoma con una struttura anormale, chiamato isocromosoma.
Pertanto, questo cromosoma tende ad avere due braccia corte invece di una di ciascuna configurazione p (corta) e lunga (q)..
Di conseguenza, la presenza di materiale genetico extra e / o anormale altererà il normale ed efficiente corso dello sviluppo fisico e cognitivo della persona affetta, dando origine alle caratteristiche cliniche della sindrome di Pallister-Killian..
La sindrome di Pallister-Killian può essere identificata durante la gravidanza o nella fase postnatale, sulla base delle caratteristiche cliniche e dei risultati di diversi test di laboratorio..
Durante la gravidanza, i test più comunemente usati sono le ecografie, l'amniocentesi o il prelievo dei villi coriali. In questo senso, l'analisi del materiale genetico dell'embrione può offrirci una conferma di questa patologia, attraverso l'identificazione di anomalie compatibili..
Se invece la diagnosi viene fatta dopo la nascita è fondamentale:
Non sono state progettate terapie specifiche per il trattamento di persone con sindrome di Pallister-Killian..
Questa sindrome è solitamente associata a una prognosi neurologica infausta e ad alti tassi di mortalità. Tuttavia, il trattamento riabilitativo, l'educazione speciale e la terapia occupazionale possono offrire una buona prognosi funzionale e un aumento della qualità della vita delle persone colpite..
Ad esempio, Méndez e il suo team di lavoro (2013) descrivono un caso di trattamento riabilitativo caratterizzato da:


Nessun utente ha ancora commentato questo articolo.