
Il Sindrome di Williams è un disturbo dello sviluppo di origine genetica che è associato a un profilo caratteristico di menomazioni fisiche e cognitive. Specificamente a livello clinico, è caratterizzato da 4 punti cardinali: 1) tratti e caratteristiche facciali atipici, 2) ritardo generalizzato nello sviluppo psicomotorio e profilo neurocognitivo specifico, 3) alterazioni cardiovascolari et) possibilità di sviluppare ipercalcemia infantile.
Nonostante il fatto che la sindrome di Williams sia considerata una malattia rara, ci sono migliaia di persone affette in tutto il mondo. Per quanto riguarda la diagnosi, l'esame clinico fornisce solitamente i rilievi necessari per la sua istituzione, tuttavia, per escludere altre patologie e falsi positivi, viene solitamente avviato uno studio genetico attraverso varie tecniche.
D'altra parte, non esiste né una cura per la sindrome di Williams né un protocollo di trattamento standard, quindi la maggior parte degli interventi terapeutici tenterà di regolare le complicanze mediche. Inoltre, sarà essenziale includere negli interventi programmi di assistenza precoce, educazione speciale personalizzata e stimolazione neuropsicologica..
Indice articolo
La sindrome di Williams è un disturbo dello sviluppo che può influenzare in modo significativo diverse aree.
Generalmente, questa patologia è caratterizzata dalla presenza di tratti del viso atipici o alterazioni cardiovascolari, disabilità intellettiva moderata, problemi di apprendimento e tratti distintivi della personalità..
Così, il primo paziente con sindrome di Williams fu descritto dal dottor Guido Fanconi, in un rapporto clinico del 1952. Fu però il cardiologo Joseph Williams che nel 1961 identificò con precisione questa patologia, oltre ad essere descritta dal tedesco Beuren.
A causa di ciò, la sindrome di Williams riceve il nome da entrambi gli autori (sindrome di Williams-Beuren), o semplicemente dal primo.
Nonostante fino a pochi anni fa l'identificazione della patologia fosse effettuata in base alle caratteristiche fenotipiche, nel 1993 Edward et al.hanno trovato un'anomalia genetica nel cromosoma 7q 11.23 come causa eziologica..
Sebbene la sindrome di Williams sia associata a un'ampia varietà di complicanze mediche secondarie, non ha un alto tasso di mortalità. In molti casi, gli individui affetti sono in grado di raggiungere un livello funzionale indipendente.
La sindrome di Williams è considerata una malattia genetica rara o rara.
La Williams Syndrome Association, tra le altre istituzioni, ha stimato che la sindrome di Williams ha una prevalenza di circa 1 caso su 10.000 persone in tutto il mondo. Nello specifico, è stato identificato che negli Stati Uniti potrebbero essercene circa 20.000 o 30.000 colpiti.
Per quanto riguarda la distribuzione della patologia per sesso, non ci sono dati recenti che indichino una maggiore prevalenza in nessuna di esse, inoltre, non sono state individuate differenze tra regioni geografiche o gruppi etnici.
D'altra parte, sappiamo anche che la sindrome di Williams è una condizione medica sporadica, sebbene siano stati descritti alcuni casi di trasmissione familiare.
La sindrome di Williams, come altre patologie di origine genetica, ha un decorso clinico caratterizzato da coinvolgimento multisistemico.
Molti autori, come González Fernández e Uyaguari Quezada, descrivono lo spettro clinico della sindrome di Williams suddiviso in diverse aree: caratteristiche biomediche, caratteristiche psicomotorie e cognitive, caratteristiche psicologiche e comportamentali, tra le altre..
L'affettività fisica presente nella sindrome di Wiliams è diversa, tra i reperti clinici più frequenti possiamo osservare:
Lo sviluppo ritardato o rallentato può già essere rilevato durante la gravidanza. I bambini affetti dalla sindrome di Williams nascono spesso con basso peso e altezza. Inoltre, una volta raggiunto lo stadio adulto, l'altezza totale è solitamente inferiore a quella della popolazione generale, circa 10-15 cm.
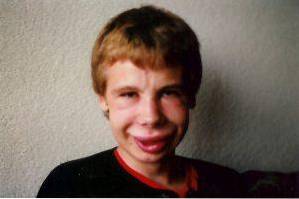
Le alterazioni facciali sono uno dei reperti clinici più caratteristici di questa sindrome. Negli individui affetti possiamo osservare una fronte significativamente stretta, pieghe cutanee marcate nella fessura palpebrale, strabismo, iride stellata, naso corto e appiattito, zigomi prominenti e un mento più piccolo del solito..
In caso di alterazioni legate allo sviluppo di muscoli e ossa, è possibile osservare la presenza di riduzione del tono e della forza muscolare, lassità articolare, scoliosi, contratture, tra gli altri. Visivamente si può osservare una postura caratterizzata da spalle cadenti e estremità inferiori semiflesse..
Sebbene di solito non vi siano anomalie o malformazioni significative nel padiglione auricolare, in tutti i casi si sviluppa un aumento della sensibilità uditiva. Gli individui affetti tendono a percepire o sperimentare certi suoni come fastidiosi o dolorosi.
La pelle di solito ha poca elasticità, quindi è possibile osservare i primi segni di invecchiamento. Inoltre, possono svilupparsi ernie, specialmente nella regione inguinale e ombelicale..
Le diverse anomalie del cuore e dei vasi sanguigni costituiscono la complicanza medica più significativa, poiché possono mettere in pericolo la sopravvivenza della persona colpita.
Tra le anomalie cardiovascolari, alcune delle più comuni sono la stenosi aortica sopravalvolare, la stenosi del ramo polmonare, la stenosi della valvola aortica. Tutte queste alterazioni, a livello clinico, possono interessare altri territori vascolari e persino il cervello, a causa dello sviluppo dell'ipertensione arteriosa.
Le anomalie legate alla funzione renale e alla vescica sono molto comuni. Inoltre, è possibile rilevare anche un accumulo di calcio (nefrocalcinosi), urgenza urinaria o enuresi notturna.
A livello cognitivo, le caratteristiche più significative sono costituite da un ritardo generalizzato nell'acquisizione delle capacità motorie, moderato ritardo intellettuale e varie alterazioni legate alla percezione visiva.
Vengono descritte varie alterazioni legate a problemi di equilibrio e coordinazione, che sono principalmente dovute alla presenza di anomalie muscolo-scheletriche e che causeranno, tra l'altro, un ritardo nell'acquisizione dell'andatura, capacità motorie finali, ecc..
È possibile riscontrare un moderato ritardo mentale, il QI tipico delle persone colpite oscilla solitamente tra 60 e 70. Per quanto riguarda le aree specifiche che sono colpite, c'è una netta asimmetria: oltre alla coordinazione psicomotoria, alla percezione e all'integrazione visiva, solitamente essere chiaramente influenzati, mentre aree come la lingua sono solitamente più sviluppate.
Nelle fasi più iniziali, di solito c'è un ritardo nell'acquisizione delle abilità linguistiche, tuttavia, di solito si riprende intorno ai 3-4 anni. I bambini con sindrome di Williams tendono ad avere una buona comunicazione espressiva, sono in grado di utilizzare il vocabolario contestualizzato, la grammatica corretta, il contatto visivo, le espressioni facciali, ecc..
Uno dei risultati più significativi nella sindrome di Williams è l'eccezionale comportamento sociale delle persone colpite. Sebbene in alcuni casi possano verificarsi crisi d'ansia o preoccupazioni eccessive, sono molto empatici e sensibili.
La ricerca più recente ha indicato che la causa della sindrome di Williams si trova in varie alterazioni genetiche sul cromosoma 7. I cromosomi trasportano le informazioni genetiche di ogni persona e si trovano nel nucleo delle cellule del corpo.
Nell'uomo possiamo trovare 46 cromosomi distribuiti a coppie. Questi sono numerati da 1 a 23, ad eccezione dell'ultima coppia costituita dai cromosomi sessuali, denominata XX nel caso delle donne, XY nel caso degli uomini. Quindi, all'interno di ogni cromosoma può esserci un'infinità di geni.
Nello specifico, il processo anormale identificato nella sindrome di Williams è una microcelezione o rottura di una molecola di DNA che conferma questo cromosoma. Normalmente, questo tipo di errore si verifica nella fase di sviluppo dei gameti maschili o femminili..
Anomalie genetiche si riscontrano nell'area 7q11.23, in cui sono stati identificati più di 25 geni differenti legati al pattern clinico caratteristico di questa patologia..
Alcuni dei geni, come Clip2, ELN, GTF21, GTF2IRD1 o LIMK1, sono assenti nelle persone colpite. La perdita di ELN è correlata ad anomalie del tessuto connettivo, della pelle e cardiovascolare.
D'altra parte, alcune ricerche indicano che la perdita dei geni Clip2, GTF2I, GTF2IRD1 e LIMK1 può spiegare alterazioni nei processi visuoperceptivi, fenotipo comportamentale o deficit cognitivi.
Inoltre, in particolare, il gene GTF2IRD1 sembra giocare un ruolo di primo piano nello sviluppo di caratteristiche facciali atipiche. Da parte sua, il gene NCF1 sembra essere correlato ad un alto rischio di sviluppare ipertensione.
Fino agli ultimi anni, la diagnosi della sindrome di Williams era basata esclusivamente sull'osservazione delle caratteristiche fenotipiche (alterazioni facciali, disabilità intellettiva, deficit cognitivi specifici, tra gli altri).
Tuttavia, attualmente, la diagnosi della sindrome di Williams viene solitamente effettuata in due fasi: analisi dei risultati clinici e studi genetici di conferma. Pertanto, la diagnosi clinica di solito include:
- Esame e valutazione fisico e neurologico.
- Analisi dei parametri di crescita.
- Esame del sistema cardiorespiratorio.
- Esame nefrourologico.
- Analisi dei livelli di calcio nelle urine e nel sangue.
- Analisi oftalmologica.
D'altra parte, l'analisi genetica viene utilizzata per confermare la presenza di alterazioni genetiche compatibili con la sindrome di Williams, tra i test più comuni c'è la tecnica di ibridazione in situ fluorescente (FIHS)..
Dopo l'estrazione di un campione di sangue, la tecnica di ibridazione in situ viene eseguita marcando le sonde di DNA che vengono rilevate sotto una luce fluorescente..
Non esiste un trattamento specifico per la sindrome di Williams, tuttavia, questa patologia è associata a più complicazioni in diversi organi, quindi gli interventi medici saranno orientati al trattamento di questi.
Gli autori González Fernández e Uyaguari Quezada sottolineano che tutti gli interventi devono avere una marcata natura multidisciplinare, consentendo il trattamento della varietà sintomatica caratteristica di questa sindrome. Inoltre, segnalano anche varie misure terapeutiche a seconda dell'area interessata:
In questo caso, complicazioni mediche come alterazioni cardiache o malformazioni muscolo-scheletriche richiedono solitamente un trattamento basato principalmente sulla somministrazione di farmaci e procedure chirurgiche. I professionisti medici di diverse aree (pediatri, cardiologi, oftalmologi, ecc.) Di solito partecipano al trattamento dei sintomi fisici..
I deficit cognitivi come l'alterazione visivo-percettiva o il ritardo linguistico dovrebbero essere affrontati sin dalle prime fasi. La stimolazione cognitiva e la riabilitazione saranno un fattore determinante per il raggiungimento di una vita autonoma durante l'età adulta.
Sebbene le persone affette dalla sindrome di Williams di solito presentino un buon funzionamento sociale, in alcune occasioni tendono a mostrare comportamenti eccessivamente ansiosi e sviluppare comportamenti perseveranti o fobie.
Pertanto, in questi casi sarà fondamentale implementare un approccio psicologico, attraverso varie strategie che siano efficaci per minimizzare questi problemi o difficoltà..



Nessun utente ha ancora commentato questo articolo.