
Il Sindrome di DiGeorge è una patologia di origine genetica che si manifesta con lo sviluppo di malformazioni legate alla struttura del cuore, del viso, del timo e delle ghiandole paratiroidi.
A livello clinico, produrranno un'ampia varietà di complicanze mediche, tra cui immunodeficienze, ipocalcemia, patologie cardiache e disturbi psichiatrici..

Per quanto riguarda l'origine eziologica, è associata ad un'alterazione genetica del cromosoma 22. Per questo è anche chiamata sindrome da delezione 22q11.2..
La diagnosi si basa sull'identificazione dei segni clinici cardinali attraverso l'esame obiettivo e vari test di laboratorio: esame analitico e immunologico, ecografia addominale, ecocardiogrammi e studio genetico, fondamentalmente basato sull'ibridazione in situ fluorescente (FISH)..
Infine, il trattamento di questa patologia si concentra sulla correzione delle malformazioni organiche e sul controllo delle complicanze mediche. Pertanto, vengono solitamente utilizzati terapia con linfociti T, integratori di calcio, chirurgia correttiva, ecc..
Indice articolo
Questa patologia è stata inizialmente descritta dallo specialista pediatrico americano Angelo M. DiGeorge nel 1965. Nel suo rapporto clinico DiGeroge ha descritto una patologia congenita definita dallo sviluppo carente o dall'assenza della ghiandola paratiroidea e del timo.
Successivamente, Chapelle nel 1918, descrisse specificamente i difetti congeniti derivati da questa patologia. Pertanto, la sindrome di DiGeorge è stata indicata come la seconda causa di difetti cardiaci congeniti dopo la sindrome di Down.
Infine, questa patologia è stata caratterizzata clinicamente attraverso la classica triade di immunodeficienza, endocrinopatia con ipocalcemia e cardiopatia..
Inoltre, in molti casi, l'ampia eterogeneità sintomatica delle delezioni localizzate sul cromosoma 22 implica la differenziazione di tre diversi tipi di patologie a livello clinico:
- Sindrome di DiGeorge
- Sindrome velocardiofacciale
- Sindrome cardiofacciale
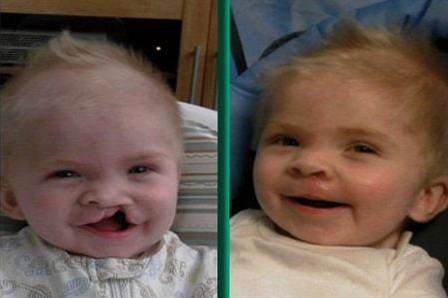
La sindrome di DiGeorge, nota anche come sindrome da delezione 22q11.2, è una malattia causata da un difetto genetico che provoca lo sviluppo di varie malformazioni organiche e corporee.
In questo senso, questa sindrome deriva fondamentalmente da processi di sviluppo difettosi durante la fase prenatale o di gestazione, localizzati principalmente durante la 3a e 8a settimana di gestazione..
Nello specifico, intorno alla 5a settimana di gestazione, le strutture embrionali iniziano un processo di formazione e sviluppo di diverse strutture e organi (Vera de Pedro et al., 2007).
Quindi, un gruppo di determinate cellule darà origine allo sviluppo del viso, di varie parti del cervello, del timo, del cuore, dell'aorta e delle ghiandole paratiroidi..
Questo "campo di cellule" si trova solitamente intorno all'area o all'area dietro il collo dell'embrione durante la gestazione. In questo modo, affinché il resto delle strutture inizi a formarsi e differenziarsi, è essenziale che queste cellule si muovano verso le diverse aree specifiche per ciascuna struttura..
In questa fase di sviluppo si formano le borse, gli archi e le fessure faringee, il timo e le ghiandole paratiroidi e, successivamente, parte delle strutture craniche e facciali o varie porzioni del tessuto connettivo.
In questo modo, le anomalie genetiche tipiche della sindrome di DiGeroge danno luogo ad un'alterazione sistematica di questo processo di formazione prenatale, provocando gravi fallimenti evolutivi.
Di conseguenza, le aree più colpite sono solitamente:
- Cuore: questa struttura costituisce uno degli organi vitali per la nostra sopravvivenza. Fa parte del sistema circolatorio e la sua funzione essenziale è pompare il sangue al resto del corpo.
- Configurazione facciale: la formazione della struttura facciale dipende dalla corretta formazione del cranio, dei bulbi oculari, del sistema buccale, delle orecchie, ecc..
- Truffa: questa struttura gioca un ruolo fondamentale all'interno del sistema immunitario, poiché è responsabile della maturazione dei linfociti o dei linfociti T.
- Ghiandole paratiroidi: sono costituiti da un insieme di ghiandole endocrine che hanno un ruolo importante nella regolazione del calcio, tra gli altri fattori.
Pertanto, le aree più colpite nella sindrome di DiGeorge sono legate al difetto di formazione embrionale nelle aree associate al collo e alle regioni adiacenti..
La sindrome di DiGeroge ha una prevalenza stimata di 1 caso ogni 4.000 persone nella popolazione generale.
Tuttavia, numerosi studi epidemiologici indicano una maggiore prevalenza principalmente a causa dell'eterogeneità del suo decorso clinico e della difficoltà di stabilire una diagnosi precoce.
Inoltre, sia negli Stati Uniti che a livello internazionale, la sindrome di DiGeorge è considerata una delle cause più comuni di difetti cardiaci congeniti e malformazioni facciali..
Per contro, in termini di caratteristiche epidemiologiche di natura sociodemografica, è stata individuata una prevalenza di 1 caso ogni 6.000 persone di origine caucasica, asiatica e afro-discendente, mentre nel caso degli ispanici la prevalenza è pari a un caso per ogni 3.800 individui.
Nel caso dei segni e dei sintomi più frequenti nella sindrome di DiGeorge, bisogna sottolineare che presenta un decorso clinico con espressività variabile.
In questo caso, in alcuni pazienti le complicanze mediche presentano uno stato grave, che può portare a morte prematura. In altri casi, le caratteristiche presentano solitamente un minimo compromesso per la sopravvivenza e la funzionalità della persona interessata..
Pertanto, non tutte le persone affette dalla sindrome di Di George presenteranno la stessa affettazione, tuttavia di solito includono una o più alterazioni correlate.
Le alterazioni legate alla configurazione facciale costituiscono una delle caratteristiche visive più eclatanti della sindrome di DiGeorge, generalmente queste sono definite da:
- Microcefalia: la testa si sviluppa con una dimensione minore o minore di quella prevista per il livello di sviluppo e l'età cronologica della persona interessata. Inoltre, una struttura nasale tubolare si sviluppa solitamente accompagnata da guance piatte o scarsamente accentuate.
- Iploplasia mandibolare e retrognazia: la struttura della mascella non è completamente sviluppata. Pertanto, in molti casi ha una dimensione ridotta o una posizione alterata, situata più indietro del solito..
- Alterazione oculare: generalmente gli occhi tendono ad essere inclusi verso il piano inferiore, inoltre, intorno agli occhi possono comparire microftalmia (sottosviluppo di uno dei bulbi oculari), cataratta (opacità del cristallino) o cianosi (colorazione blu).
- Alterazione del padiglione auricolare: è possibile identificare un'asimmetria nella configurazione delle orecchie. Di solito hanno un impianto basso con presenza di malformazioni nei lobi e in altre aree esterne del padiglione auricolare.
- Malformazioni orali: la configurazione della bocca presenta solitamente un aspetto arcuato verso il piano superiore, caratterizzato dalla presenza di un lungo e accentuato solco naso-labiale e palatoschisi.
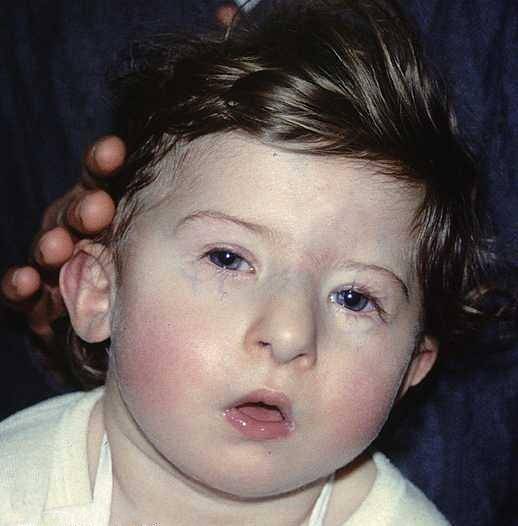
Le anomalie cardiache spesso includono un'ampia varietà di difetti. Tuttavia, le aree più colpite sono legate all'aorta e alle strutture cardiache associate:
- Difetti settali: il muro o la struttura che separa le camere cardiache responsabili del pompaggio del sangue, può essere formato in modo incompleto o difettoso.
- Malformazione dell'arco aortico: Diverse anomalie possono essere descritte anche nel segmento aortico situato tra le vie ascendenti e discendenti.
- Tetralogia di Fallot: questa patologia si riferisce alla presenza di alterazioni del difetto del setto ventricolare, restringimento significativo dell'arteria polmonare, posizione anormale dell'aorta e ispessimento dell'area ventricolare destra.
Le persone affette dalla sindrome di DiGeorge hanno solitamente una notevole suscettibilità a contrarre vari tipi di patologie, principalmente infettive (virus, funghi, batteri, ecc.).
Questo fatto è dovuto alla presenza di una disfunzione del sistema immunitario, a causa di uno sviluppo carente del tipo e della produzione di linfociti e cellule T.
Il sistema immunitario è costituito da un'ampia varietà di organi, strutture, tessuti e cellule che insieme ci proteggono da agenti patologici ambientali e interni..
In questo senso, la sindrome di DiGeorge produce una formazione carente o incompleta del timo, portando ad alterazioni della sua funzionalità e localizzazione finale..
Generalmente, l'anomalia più evidente è l'ipofunzionalità dei linfociti T, essenziale nella produzione di immunoglobuline e anticorpi..
In questo caso, le persone affette dalla sindrome di Digeorge di solito hanno livelli anormalmente bassi di concentrazione di calcio nel corpo e nel flusso sanguigno..
Questa condizione medica deriva fondamentalmente dalla presenza di anomalie nelle ghiandole paratiroidi, a causa di un sottosviluppo dei suoi componenti (PrimaryInmune, 2011).
Queste ghiandole si trovano nel collo e sono in una posizione vicino alla tiroide. Tuttavia, in questo caso hanno un volume ridotto, quindi avrà un impatto significativo sul controllo del metabolismo e sull'equilibrio del calcio nel corpo..
Pertanto, in questo caso, il livello di calcio nel sangue è solitamente inferiore a 2,1-8,5 mm / dl, causando diverse complicazioni mediche come crampi, irritabilità muscolare, intorpidimento, sbalzi d'umore, deficit cognitivi, ecc..
Oltre ai segni e sintomi sopra descritti, è possibile individuarne altri legati alla sfera cognitiva e intellettuale di chi ne è affetto..
Soprattutto nei casi diagnosticati, sono state descritte difficoltà di apprendimento, deficit cognitivo moderato, deficit di attenzione, alterazioni dell'umore, disturbi d'ansia, tra gli altri..
L'origine genetica della sindrome di DiGeorge è associata alla presenza di alterazioni nel cromosoma 22, in particolare nella posizione 22q11.2. In particolare, è dovuto all'assenza di una sequenza di DNA, composta da un numero compreso tra 30 e 40 geni diversi..
Sebbene una buona parte dei geni coinvolti non sia stata ancora identificata in dettaglio, l'assenza di questo ampio gruppo si verifica in più del 90% dei casi come mutazione de novo, mentre circa il 7% è dovuta a fattori ereditari.
Per l'istituzione della diagnosi della sindrome di DiGeorge, è essenziale identificare i segni clinici cardinali di questa patologia:
- Difetti facciali.
- Difetti cardiaci.
- Immunodeficienza.
- Ipocalcemia.
In questo senso, insieme all'analisi dell'anamnesi e all'esame obiettivo, è fondamentale eseguire vari esami di laboratorio come l'ecocardiografia, l'ecografia, l'esame immunologico e gli studi analitici del siero..
Inoltre, un aspetto importante è l'esame genetico, questo viene effettuato principalmente tramite ibridazione in situ fluorescente (FISH).
Come abbiamo sottolineato nella descrizione iniziale, il trattamento è principalmente finalizzato al controllo e alla correzione dei segni e dei sintomi causati da questo tipo di malattia..
In caso di ipocalcemia, viene solitamente trattata mediante la somministrazione di integratori di calcio e / o vitamina D..
D'altra parte, in caso di immunodeficienza, sebbene tendano a migliorare con l'età, possono essere utilizzati vari approcci, come il trapianto di parte del tessuto del timo, la terapia con linfociti T o il trapianto di midollo osseo..
Per quanto riguarda le malformazioni facciali e orali, vengono solitamente utilizzate riparazioni chirurgiche, che migliorano l'aspetto fisico e la funzionalità di queste ossa.
Infine, in caso di alterazioni cardiache, entrambi i farmaci possono essere somministrati per il loro trattamento e correzione mediante intervento chirurgico..
Nella maggior parte dei casi, le persone colpite di solito raggiungono l'età adulta, tuttavia, una percentuale significativa di queste inizia a sviluppare importanti anomalie immunologiche e / o cardiache che causano morte prematura, specialmente entro il primo anno di vita..



Nessun utente ha ancora commentato questo articolo.