
Il Sindrome di Apert o acrocefalosindattilia di tipo I (ACS1) è una patologia di origine genetica che si caratterizza per la presenza di diverse alterazioni e malformazioni nel cranio, nel viso e nelle estremità.
A livello clinico, la sindrome di Apert è caratterizzata dalla presenza o dallo sviluppo di un cranio appuntito o allungato, area facciale infossata con alterazione nella proiezione dei denti, fusione e chiusura delle ossa e delle articolazioni delle dita, ritardo mentale variabile, disturbi del linguaggio , eccetera..
Sebbene questa patologia possa essere ereditaria, nella maggior parte dei casi la sindrome di Apert si manifesta senza la presenza di una storia familiare, essenzialmente a causa di una mutazione de novo durante la fase di gestazione..
I meccanismi genetici che causano la sindrome di Apert non sono esattamente noti. Attualmente sono state individuate varie alterazioni genetiche che sono in grado di produrre questa patologia, essenzialmente legate a mutazioni nel gene FGFR2.
D'altra parte, la diagnosi di sindrome di Apert di solito inizia con sospetto clinico nel periodo prenatale dopo l'identificazione di anomalie nelle scansioni ecografiche di routine ed è confermata conducendo uno studio genetico.
Per quanto riguarda il trattamento, non esiste alcun tipo di intervento curativo per la sindrome di Apert. Tuttavia, nel corso della storia di questa patologia, sono stati progettati vari interventi specifici che di solito includono neurochirurgia, chirurgia cranio-facciale, chirurgia maxillo-facciale, trattamento farmacologico, terapia fisica, intervento psicologico e neuropsicologico, tra gli altri.
Indice articolo
La sindrome di Apert è una patologia genetica caratterizzata dalla presenza di diverse malformazioni scheletriche a livello cranico, facciale e / o degli arti.
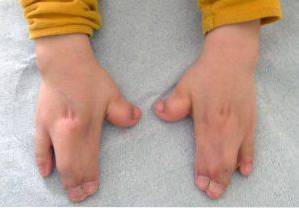
L'alterazione essenziale della sindrome di Apert è costituita da una chiusura prematura o precoce delle fessure craniche, che provoca una crescita anormale del resto delle strutture del viso e del cranio. Oltre a queste, possono comparire malformazioni anche negli arti superiori e inferiori, come la fusione delle dita delle mani e dei piedi..
D'altra parte, possono essere influenzate anche le capacità cognitive delle persone affette da sindrome di Apert, con una gravità variabile da lieve a moderata..
Sebbene Baumgartner (1842) e Wheaton (1894) abbiano fatto le prime menzioni su questa condizione medica, fu solo nel 1906, quando il medico specialista francese Eugene Apert, descrisse accuratamente questa sindrome e pubblicò il primo rapporto clinico..
Nella sua pubblicazione, Eugene Apert, descrive una serie di nuovi casi di pazienti affetti da un pattern malformativo ben definito e caratterizzati dai segni e sintomi caratteristici di questa patologia..
Pertanto, non è stato fino al 1995 che sono stati identificati i fattori genetici eziologici della sindrome di Apert. Nello specifico, Wilkie et al.Hanno descritto la presenza di due mutazioni nel gene FGFR2 in circa 40 pazienti affetti..
Inoltre, la sindrome di Apert è una condizione medica classificata tra le malattie o patologie caratterizzate dalla presentazione di craniosinostosi (chiusura prematura delle suture craniche).
Altre patologie appartenenti a questo gruppo sono la sindrome di Pfeiffer, la sindrome di Crouzon, la sindrome di Saethre-Chotzcen e la sindrome di Carpenter..
La sindrome di Apert è considerata una patologia rara o poco frequente, cioè ha una prevalenza inferiore a un caso ogni 15.000 abitanti della popolazione generale..
Nello specifico, la sindrome di Apert si manifesta intorno a una persona ogni 160.000-200.000 nascite e, inoltre, c'è una probabilità del 50% di trasmettere questa patologia a livello ereditario.
Inoltre, per quanto riguarda la distribuzione per sesso, non è stata individuata una maggiore prevalenza negli uomini o nelle donne, né è stata associata a particolari gruppi etnici o aree geografiche..
Attualmente, e dato che la sindrome di Apert è stata identificata intorno al 1984, nelle relazioni cliniche e nella letteratura medica che hanno pubblicato più di 300 casi di questa patologia.
Le manifestazioni cliniche della sindrome di Apert di solito includono una malformazione o uno sviluppo incompleto della struttura cranica, un fenotipo atipico o un pattern facciale e alterazioni scheletriche alle estremità..
Nel caso della sindrome di Apert, il coinvolgimento centrale è legato alla formazione e chiusura della struttura ossea del cranio. Durante lo sviluppo embrionale si verifica un processo chiamato creneosinostosi, caratterizzato dalla chiusura prematura delle suture craniche.
Le fessure o suture craniche sono un tipo di bande di tessuto fibroso che hanno l'obiettivo fondamentale di collegare le ossa che compongono il cranio (frontale, occipitale, parietale e temporale).
Durante la fase di gestazione e il primo periodo postnatale, la struttura ossea che compone il cranio è tenuta insieme grazie a questi tessuti fibrosi ed elastici.
Normalmente, le ossa craniche non si fondono fino a circa 12-18 mesi. La presenza di punti molli o spazi tra le ossa craniche fa parte del normale sviluppo del bambino.
Pertanto, durante l'intera fase dell'infanzia, queste suture o regioni flessibili consentono al cervello di crescere in modo accelerato e, inoltre, lo proteggono dagli impatti.
Pertanto, nella sindrome di Apert, la chiusura prematura di queste suture craniche e delle ossa craniche rende impossibile il normale sviluppo della crescita cranica e cerebrale..
Di conseguenza, i segni e sintomi più comuni della sindrome di Apert possono includere:
Questi tipi di alterazioni interessano principalmente gli arti superiori e inferiori, normalmente la fusione e lo sviluppo delle dita..
Oltre a questi, è possibile osservare anche altri reperti clinici a livello muscolo-scheletrico, accorciamento di varie ossa (radio, omero, femore), ipoplasia della scapola o del bacino, fusione delle vertebre cervicali.
Di conseguenza, molte persone affette presenteranno una ridotta mobilità articolare e, quindi, potrebbero sviluppare varie difficoltà per l'acquisizione delle capacità motorie grossolane e fini.
Questi tipi di anomalie sono molto eterogenei e variabili tra gli individui affetti, tuttavia, alcuni dei più comuni sono stati identificati:
L'alterazione eziologica di questa patologia può portare allo sviluppo di lesioni o patologie secondarie a livello morfologico e strutturale in varie zone del corpo, alcune delle quali comprendono:
Nonostante in molti casi sia possibile osservare la presenza di un'alterazione generale delle funzioni cognitive e del livello intellettuale, il Ritardo mentale non è inequivocabilmente presente in tutti i casi di sindrome di Apert..
Inoltre, nei casi in cui è presente una compromissione del livello intellettuale, questa può essere variabile, su una scala da lieve a moderata.
In ambito linguistico, invece, è frequente lo sviluppo di vari deficit, principalmente legati all'articolazione dei suoni a seguito di malformazioni mandibolari e orali..
La sindrome di Apert è dovuta alla presenza di una specifica mutazione nel gene FGFR2. Studi sperimentali hanno indicato che questo gene è responsabile della produzione di una proteina, chiamata recettore 2 del fattore di crescita dei fibroblasti.
Tra le funzioni di questo fattore, descrive l'invio di diversi segnali chimici a cellule immature per provocare la loro trasformazione e differenziazione in cellule ossee durante la fase di sviluppo fetale o prenatale..
Pertanto, la presenza di mutazioni nel gene FGFR2 altera il funzionamento di questa proteina e, quindi, può causare la fusione precoce delle ossa del cranio, delle mani e dei piedi..
Una buona parte delle caratteristiche cliniche della sindrome di Apert può essere identificata durante la gravidanza, in particolare negli esami ecografici della gravidanza e dello sviluppo fetale..
Pertanto, in caso di sospetto clinico, viene riavviato uno studio genetico per identificare la presenza di una mutazione genetica compatibile con la sindrome di Apert..
Quando invece i segni sono impercettibili o non sono stati identificati prima della nascita, dopo di ciò è possibile eseguire un'analisi fisica dettagliata e vari test genetici per confermare la diagnosi..
Sebbene non esista una cura specifica per la sindrome di Apert, sono stati descritti vari approcci per il trattamento dei sintomi e delle complicanze mediche di questa patologia..
Gli interventi terapeutici più efficaci sono quelli che vengono attuati precocemente, nei primi momenti di vita e coinvolgono professionisti di diversi ambiti.
In genere, il trattamento dei bambini affetti richiede una pianificazione personalizzata, con più interventi chirurgici programmati. Pertanto, la gestione di questa patologia si basa sulla correzione delle malformazioni scheletriche e cranio-facciali e sul supporto psicologico e neuropsicologico..
Attraverso la neurochirurgia l'obiettivo è ricostruire la volta cranica, mentre gli specialisti in chirurgia maxillo-facciale cercano di correggere le malformazioni facciali. Frequente, invece, è anche la partecipazione di chirurghi traumatologici, per la ricostruzione delle malformazioni presenti nelle mani e nei piedi.
Inoltre, la progettazione di programmi individualizzati per la stimolazione precoce, la riabilitazione della comunicazione, la formazione delle abilità sociali o il follow-up psico-pedagogico, sono utili per il raggiungimento di uno sviluppo ottimale, funzionale e indipendente degli individui colpiti..


Nessun utente ha ancora commentato questo articolo.