Il prioni sono proteine senza un genoma o acidi nucleici che agiscono come agenti infettivi. Il termine "prione" significa particella infettiva proteica (dall'inglese Proteinaceous Infectious Particles) ed è stato coniato dal neurologo e vincitore del Premio Nobel Stanley B. Prusiner.
Nel 1982, Prusiner ei suoi colleghi hanno identificato una particella proteica infettiva, mentre studiavano le cause delle malattie di Creutzfeldt-Jakob (nell'uomo) e dell'encefalopatia spongiforme bovina..
Questi rari agenti infettivi si trovano nella membrana delle cellule normali, solo come proteine mal ripiegate e / o con una struttura tridimensionale anormale. Queste proteine sono responsabili di molteplici malattie degenerative e di altissima mortalità che colpiscono i tessuti neurali e la struttura del cervello..
Sono anche chiamate malattie da prioni. Tra i più importanti che colpiscono gli esseri umani ci sono il kuru, la malattia di Gerstmann-Sträussler-Scheinker, la sindrome di Creutzfeldt-Jakob e l'insonnia familiare fatale..
Indice articolo
I prioni sono strutture proteiche presenti nelle membrane cellulari. Queste proteine hanno una forma o conformazione alterata [PrP (Sc)].
Per quanto riguarda la sua moltiplicazione, si ottiene mediante la conversione delle forme, come nel caso della scrapie. In questa malattia, i prioni reclutano PrP (C) (proteine prioniche di conformazione inalterata) per stimolare la conversione nell'isoforma PrP (Sc)..
Questo genera una reazione a catena che diffonde il materiale infettivo e quindi permette l'irrigazione della malattia. Non è ancora noto come avvenga questo processo di conversione.
Queste proteine insolite in grado di propagarsi, non hanno acidi nucleici. Prova di ciò è che sono resistenti ai raggi X e alle radiazioni ultraviolette. Questi agenti scompongono facilmente gli acidi nucleici.
Le proteine prioniche, di cui sono composti i prioni (PrP), si trovano in tutto il corpo, non solo negli esseri umani ma in altri vertebrati sani. Queste proteine sono generalmente resistenti alle proteasi (enzimi che catalizzano le proteine).
Si sa molto poco sull'utilità delle proteine prioniche PrP (C), la forma normale della proteina non infettiva nel corpo umano..
Tuttavia, alcuni ricercatori sono riusciti a dimostrare che, nei topi, queste proteine attivano la riparazione della mielina nelle cellule del sistema nervoso periferico. L'assenza di questi ha anche dimostrato di causare la demielinizzazione di tali cellule nervose..
La conoscenza che si ha sulla struttura dei prioni risiede principalmente nelle indagini svolte sul batterio Escherichia coli.
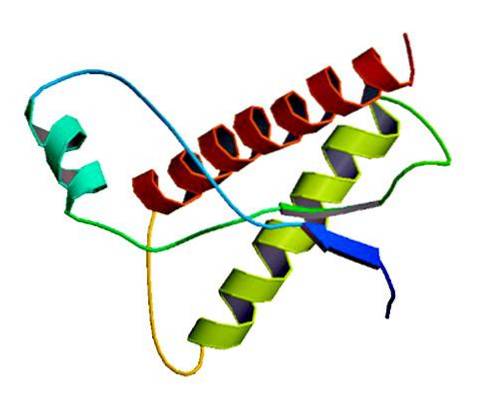
Gli studi hanno dimostrato che i polipeptidi a catena PrP (C) (normale) e PrP (Sc) (infettivo) sono identici nella composizione degli amminoacidi, ma differiscono nella loro conformazione 3D e ripiegamento..
Questi prioni non infettivi hanno 209 amminoacidi nell'uomo. Hanno un legame disolfuro. La sua struttura è alfa-elicoidale, il che significa che ha amminoacidi a forma di spirale (alfa eliche) e pochi fili piatti di amminoacidi (fogli beta)..
Questa proteina non può essere separata per centrifugazione, il che implica che non è sedimentabile. È facilmente digeribile dalla serina proteasi ad ampio spettro chiamata proteinasi K..
È una proteina infettiva che trasforma la PrP (C) in isoforme infettive PrP (Sc) e con una configurazione o forma anormale.
Si sa molto poco della sua struttura 3D, tuttavia è noto che ha poche forme elicoidali e più fili piatti o fogli beta. Il passaggio all'isoforma è ciò che è noto come l'evento cardine delle malattie da prioni.
Le proteine prioniche cellulari [Prp (C)] si trovano sulla superficie cellulare di un'ampia varietà di organi e tessuti. Si sa molto poco sulle funzioni fisiologiche dei prioni nel corpo. Anche così, gli esperimenti condotti sui topi indicano possibili funzioni, come:
È stato dimostrato che PrP (C) agisce con i recettori del glutammato (ionotropici e metabotropici). PrP (C) partecipa come recettore per gli oligomeri sinaptotossici del peptide di superficie cellulare Aβ.
Nei topi della famiglia Murinae, è stato scoperto che le proteine prioniche PrP (C) vengono espresse entro pochi giorni dall'impianto, nello sviluppo embrionale.
Ciò indica che svolgono un ruolo durante lo sviluppo di questi piccoli mammiferi. Ruolo che secondo i ricercatori è legato alla regolazione della neuritogenesi (produzione di assoni e dendriti di neuroni).
Agiscono anche sulla crescita assonale. Queste proteine prioniche sono anche coinvolte nello sviluppo del circuito cerebellare. Per questo motivo, si ritiene che l'assenza di questi prioni PrP (C) comporti un ritardo nello sviluppo motorio dei roditori..
Negli studi sulla sovraespressione della PrP (C) da parte dell'orientamento genico, è stato riscontrato che l'assenza di questi prioni causa problemi di afflusso di sangue ad alcune parti del cervello (ischemia cerebrale acuta).
Ciò significa che le proteine prioniche funzionano come neuroprotettori. Inoltre, è stato dimostrato che la sovraespressione di PrP (C) può ridurre o migliorare le lesioni causate dall'ischemia..
Recentemente è stato scoperto il ruolo fisiologico di Prp (C) nel mantenimento della mielina periferica.
Durante uno studio di laboratorio è stato scoperto che in assenza della proteina prionica, i topi di laboratorio hanno sviluppato carenze nei nervi che trasportano le informazioni dal cervello e dal midollo spinale, in quella che viene chiamata una neuropatia periferica.
Esistono alcune proteine simili ai prioni e si trovano in altre parti del corpo oltre al cervello.
La funzione di tali proteine è quella di avviare, regolare e / o controllare la morte cellulare, quando l'organismo viene attaccato (ad esempio dai vironi), prevenendo così la diffusione del patogeno..
Questa peculiare funzione di queste proteine fa riflettere i ricercatori sulla possibile importanza dei prioni non infettivi nella lotta contro i patogeni..
Uno studio condotto presso lo Stowers Institute, nel Missouri, USA ha dimostrato che i prioni PrP possono avere un ruolo nel mantenimento della memoria a lungo termine.
Lo studio ha rivelato che alcune proteine prioniche possono essere controllate per lavorare nel mantenimento delle funzioni fisiologiche della memoria a lungo termine..
Un'indagine sulle proteine prioniche espresse nelle cellule staminali del tessuto sanguigno, ha rivelato che tutte queste cellule staminali (ematopoietiche) esprimono proteine prioniche nella loro membrana cellulare. Per quello che si crede che partecipino al complesso e importantissimo processo di rinnovamento cellulare.
Le patologie di origine prionica sono riconosciute come disturbi cerebrali degenerativi progressivi. Possono attaccare bovini, cervi, caribù, pecore e persino umani.
Queste malattie sono causate da un'alterazione nella struttura delle proteine PrP (C) e le cui funzioni specifiche sono ancora oggi incerte. Le patologie da prioni possono insorgere senza una causa nota. Possono avere un'origine genetica ereditaria e possono anche essere trasmesse in modo infettivo-contagioso.
I prioni causano malattie familiari, sporadiche e contagiose. Le malattie da prioni familiari sono quelle ereditabili. Le patologie sporadiche sono le più comuni e si verificano senza cause note..
Le malattie contagiose sono considerate rare, si trasmettono da persona a persona, da animale ad animale, da persona ad animale e viceversa. Le cause sono molteplici e vanno dal consumo di carne contaminata, cannibalismo, trasfusioni, alla manipolazione di attrezzature chirurgiche contaminate.
Le malattie da prioni più comuni sono:
Considerata la malattia da prioni più comune tra gli esseri umani, è una malattia cosmopolita, cioè ha una distribuzione mondiale. Può essere ereditario (familiare), sporadico o infettivo.
I pazienti presentano sintomi come demenza, spasmi o movimenti involontari improvvisi e carenze del sistema nervoso centrale.
A seconda del trattamento e della forma della malattia, la morte può verificarsi tra 4 mesi e 2 anni dopo l'acquisizione della malattia. La diagnosi è difficile da fare, di solito viene eseguita post morten, durante l'autopsia.
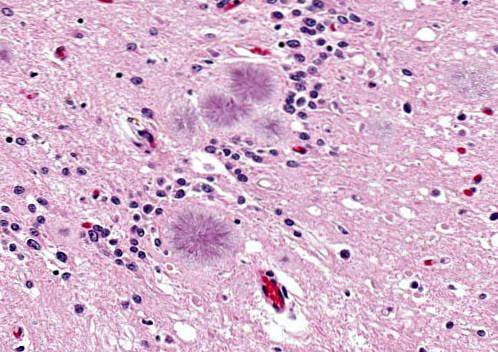
È una malattia causata dai prioni in un processo cerebrale infettivo ereditario o autosomico dominante. La malattia si manifesta in persone di età compresa tra 40 e 60 anni.
Queste persone manifestano problemi di articolazione delle parole (disartria), sussulti o movimenti involontari improvvisi, essendo frequente l'aggressività.
Si presentano con degenerazione cerebellare accompagnata da un'andatura instabile. È anche possibile osservare iporeflessia, sordità, paralisi dello sguardo, demenza, tra gli altri sintomi. L'aspettativa di vita è di circa 5 anni o un po 'più lunga.
È una malattia molto rara, al punto che il suo range di insorgenza va da 2 a 3 casi ogni 100 milioni di abitanti. La patologia è simile alla malattia di Gerstmann-Sträussler-Scheinker.
Le manifestazioni cliniche della proteina indicano una bassa resistenza alle proteasi, alcune sono più e altre meno sensibili a questi enzimi.
I sintomi che i pazienti presentano sono: problemi di linguaggio e deterioramento cognitivo, perdita di neuroni nell'area in cui il cervello controlla i movimenti ed esegue la coordinazione muscolare.
La malattia è frequente nei pazienti anziani (70 anni) e il tempo di vita stimato una volta infettato è di circa 20 mesi.
È una malattia ereditaria o familiare, può verificarsi anche sporadicamente. È noto che la malattia è dovuta a una mutazione ereditaria o autosomica dominante.
I pazienti presentano sintomi come problemi cumulativi di sonno e mantenimento del sonno, demenza, deterioramento cognitivo, persino problemi di ipertensione, tachicardia, iperidrosi e altri..
L'età che colpisce è piuttosto ampia, va dai 23 ai 73 anni, tuttavia l'età media è di 40 anni. La durata della vita una volta infettati è di poco più di 6 anni.
Questa malattia da prioni è stata rilevata solo negli abitanti della Papua Nuova Guinea. È una malattia legata al cannibalismo e alla tradizione culturale del rito del pianto dei morti, dove queste persone mangiano cervello o carne umana.
Le persone portatrici della malattia di solito hanno movimenti incontrollabili e involontari in diverse parti del corpo.
Presentano tremori, perdita di controllo dei movimenti e perdita di coordinazione muscolare. L'aspettativa di vita nelle persone infette è di due anni.
Tra le patologie prodotte dai prioni negli animali c'è l'encefalopatia spongiforme bovina. Questa malattia ha causato il caos in Europa, nella salute pubblica, in quella degli animali e nell'economia dei paesi colpiti.
Altre malattie negli animali includono la scrapie, l'encefalopatia trasmissibile del visone, la malattia da deperimento cronico (nei cervi) e l'encefalopatia spongiforme felina..
Queste malattie, come quelle presentate nell'uomo, mancano di un trattamento efficace, quindi la prevenzione è essenziale, soprattutto dopo le infezioni nell'uomo che si sono verificate a seguito del consumo di carne di vacche infette..
Ad oggi, non esiste una cura nota per le malattie da prioni. Il trattamento è sintomatico. Si consiglia ai pazienti di pianificare cure palliative e si raccomandano test genetici e consulenza per i membri della famiglia.
Un'ampia varietà di farmaci è stata testata in pazienti con malattie da prioni, come antivirali, antitumorali, farmaci per malattie come il Parkinson, trattamenti per l'immunosoppressione, antibiotici, antimicotici e persino antidepressivi..
Tuttavia, attualmente non ci sono prove che indichino che alcuni di questi riducano i sintomi o migliorino la sopravvivenza dei pazienti..
I prioni sono resistenti a una varietà di cambiamenti fisici e chimici. Tuttavia, vengono utilizzate diverse tecniche per evitare la contaminazione dei pazienti con strumenti chirurgici contaminati..
Tra le tecniche più utilizzate c'è quella di sterilizzare l'apparecchiatura in autoclave a 132 ° C per un'ora e poi immergere gli strumenti in idrossido di sodio per almeno un'altra ora..
D'altra parte, l'Organizzazione mondiale della sanità (OMS) ha sviluppato misure per prevenire la diffusione delle malattie da prioni. Questa organizzazione stabilisce norme per la manipolazione di tessuti vietati o potenzialmente rischiosi come: occhi, cervello, intestino, tonsille e midollo spinale.


Nessun utente ha ancora commentato questo articolo.