Il panipopituitarismo È una condizione caratterizzata da una produzione inadeguata o assente di ormoni dall'ipofisi anteriore. Può avere manifestazioni diverse a seconda dell'età in cui compare.
Alcuni di loro sono bassa statura, bassa pressione sanguigna, vertigini, debolezza muscolare, micropene, atrofia ottica, ipoglicemia, pelle secca, affaticamento, costipazione, ecc. Tuttavia, questi sintomi dipendono dagli ormoni interessati e variano se la condizione è congenita o acquisita..

Il panipopituitarismo può avere molteplici cause. Può apparire a causa di un problema durante il periodo embrionale. Oppure, a causa di lesioni, infiammazioni o tumori in età avanzata.
Questa malattia è cronica e necessita di cure permanenti per sostituire gli ormoni mancanti. A seconda degli ormoni da carenza, il trattamento indicato sarà diverso. Questo si basa sul supporto farmacologico.
A volte i termini ipopituitarismo e panipopituitarismo sono usati in modo intercambiabile, sebbene normalmente quest'ultimo concetto si riferisca a un deficit totale di alcuni ormoni prodotti dal lobo anteriore della ghiandola pituitaria..
Indice articolo
Sia l'ipopituitarismo che il panipopituitarismo sono condizioni molto rare. Ci sono davvero pochi studi che esaminano la prevalenza di questa condizione. La maggior parte si concentra sull'ipopituitarismo in generale.
Secondo Bajo Arenas (2009), la prevalenza dell'ipopituitarismo è di 45,5 su 100.000, 4,2 nuovi casi ogni 100.000 abitanti..
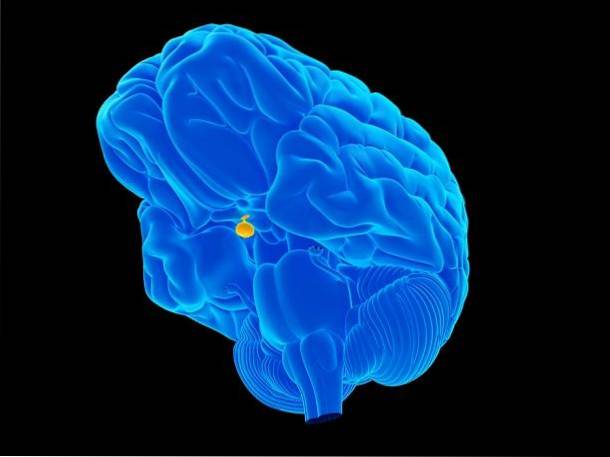
Per comprendere il panipopituitarismo, è importante conoscere la missione della ghiandola pituitaria.
La ghiandola pituitaria, chiamata anche ipofisi, è quella colpita dal panipopituitarismo. Questa ghiandola è la "maestra endocrina del corpo", poiché controlla le funzioni di altri organi endocrini..
Pertanto, secerne ormoni che regolano altre importanti ghiandole, mantenendo l'omeostasi (equilibrio) del corpo. Ricerca la corretta somministrazione di nutrienti e proteine che otteniamo dalla dieta.
Attraverso i livelli ormonali, la ghiandola pituitaria controlla funzioni come la crescita del corpo, dei capelli e delle unghie, delle mucose del corpo, del latte materno, ecc..
Questa ghiandola si trova su un osso chiamato "sella turcica" che si trova nell'osso epenoide del cranio. Grazie alla sua posizione si connette più facilmente con l'ipotalamo, attraverso una struttura chiamata peduncolo ipofisario. Le forme dell'ipotalamo controllano l'ipofisi anteriore.
L'ipofisi o ipofisi è divisa nel lobo anteriore e posteriore. Il primo produce ormone stimolante la tiroide (TSH), corticotropina, ormone luteinizzante (LH), ormone follicolo stimolante (FSH), ormone della crescita (GH) e prolattina. Mentre il successivo secerne vasopressina (ormone antidiuretico) e ossitocina.
Nel panipopituitarismo, c'è un'assenza di questi ormoni per vari motivi. Pertanto, i pazienti che ne soffrono possono avere problemi nel funzionamento del loro corpo.
Il panipopituitarismo può essere causato da cause acquisite o, meno frequentemente, da cause genetiche. Quando la ghiandola pituitaria non funziona come dovrebbe, ma le cause non sono state ancora identificate, si parla di "panipopituitarismo idiopatico"..
Le cause acquisite più comuni sembrano essere i tumori che coinvolgono la ghiandola pituitaria. La seconda causa più comune è la sindrome di Sheehan, che si verifica nelle donne dopo il parto. È caratterizzato da un infarto della ghiandola pituitaria a seguito di sanguinamento durante o dopo il parto..
Le seguenti sono le cause più possibili di panipopituitarismo:
Negli adulti, i più comuni sono gli adenomi ipofisari e rappresentano tra il 10 e il 15% dei tumori intracranici. Tendono a crescere lentamente e influenzano maggiormente le donne. Tuttavia, normalmente non metastatizzano.
Nei bambini possono verificarsi craniofaringiomi. Sono tumori che compaiono dai resti embrionali della borsa di Rathke (una struttura che durante lo sviluppo embrionale dà origine alla ghiandola pituitaria). Si manifestano con un aumento della pressione intracranica, mal di testa, vomito, bassa statura e crescita lenta.
Questa è una malattia ereditaria che colpisce il metabolismo del ferro, producendo livelli eccessivamente alti di ferro..
Malattie autoimmuni in cui il sistema immunitario fallisce e attacca i tessuti sani del corpo. Un esempio è la ghiandola pituitaria linfocitica autoimmune, in cui questo sistema distrugge i linfociti dell'ipofisi.
Problemi vascolari in questa ghiandola come la sindrome di Sheehan o l'aneurisma carotideo interno (che fornisce la ghiandola).
Si verifica quando la ghiandola pituitaria si restringe, sotto la pressione di un aumento del liquido cerebrospinale fuoriuscito.
Come mutazioni genetiche in PIT1 o PROP1. Una sindrome genetica associata al panipopituitarismo è la sindrome di Kallmann. È caratterizzato dal mancato sviluppo delle caratteristiche sessuali e delle alterazioni olfattive.
- Difetti durante lo sviluppo embrionale delle cellule che compongono l'ipofisi anteriore o ipotalamo.
- Ad esempio, lesioni infundibolari acquisite (nella parte posteriore della ghiandola pituitaria) dopo un ictus.
- Metastasi da altri tumori, come mammella, prostata, colon o polmone.
- Trattamenti di radioterapia.
- Granulomatosi (infiammazione dei vasi sanguigni) che interessa l'area dell'ipofisi o dell'ipotalamo.
- Infezioni come tubercolosi, toxoplasmosi, sifilide o micosi.
- Apoplessia ipofisaria: è un'ischemia o un'emorragia che colpisce la ghiandola pituitaria. Produce sintomi come mal di testa, vomito e deficit visivo.
- Sequele dopo un intervento chirurgico che colpisce l'ipofisi o le aree coinvolte.
- Trauma alla testa.
I sintomi del panipopituitarismo variano notevolmente a seconda delle cause, dell'età, della rapidità con cui si manifesta, degli ormoni coinvolti e del livello di gravità..
In questo modo, possono esserci pazienti che presentano ipotiroidismo grave (cattivo funzionamento della ghiandola tiroidea), mentre altri avvertono solo malessere generale o stanchezza eccessiva..
Ovviamente, le conseguenze sono peggiori quando il panipopituitarismo compare prima.
L'assenza di ormoni produce sintomi diversi a seconda di cosa sono. Pertanto, una mancanza di ormone della crescita (GH) causa bassa statura nei bambini. Mentre negli adulti porta a cambiamenti nella forma del corpo, problemi di metabolismo del glucosio e dei lipidi e malessere generale.
La carenza di gonadotropine, d'altra parte, indurrebbe una donna a ritardare o a mancare le mestruazioni e a bassa libido. Negli uomini produce disfunzione sessuale e micropene (se il problema si manifesta durante l'infanzia).
In caso di assenza di ormoni stimolatori della tiroide (TSH), invece, comparirebbe ipotiroidismo, caratterizzato da aumento di peso, affaticamento, intolleranza al freddo, dolori muscolari, stipsi, depressione, ecc..
La mancanza di ormone adrenocorticotropo o corticotropina (ACTH) ha conseguenze più negative e può mettere in pericolo la vita del paziente. Soprattutto se il deficit si verifica all'improvviso. In questo caso, si manifesta con bassa pressione sanguigna, ipoglicemia, nausea, vomito, estrema stanchezza e una bassa concentrazione di sodio nel sangue..
Se i livelli di ACTH diminuiscono lentamente, i sintomi sono perdita di peso, debolezza, affaticamento e nausea..
D'altra parte, la mancanza di prolattina è un sintomo molto significativo del panipopituitarismo. Può impedire alle donne di produrre latte dopo la gravidanza. È anche una causa della sindrome di Sheehan precedentemente descritta.
Altri sintomi generali del panipopituitarismo sono ipersensibilità al freddo, diminuzione dell'appetito, anemia, infertilità, perdita di peli pubici, mancanza di peli sul corpo, gonfiore del viso, desiderio sessuale inibito, ecc..
Possono anche manifestarsi sete eccessiva e un aumento esagerato della secrezione urinaria, che proviene dal diabete insipido. Quest'ultima condizione deriva da un deficit di vasopressina, un ormone che viene prodotto nell'ipotalamo e immagazzinato nella ghiandola pituitaria..
Il trattamento principale per il panipopituitarismo consiste nel sostituire quegli ormoni che sono assenti o carenti. Allo stesso tempo, viene trattata la causa sottostante che ha causato questa condizione.
Le dosi esatte di ormoni devono essere prescritte da un endocrinologo dopo aver eseguito le opportune analisi. Dovrebbero essere le quantità che il corpo produrrebbe naturalmente se non ci fosse il panipopituitarismo. Questa sostituzione ormonale può durare una vita.
I corticosteroidi come l'idrocortisone o il prednisone sono spesso prescritti per sostituire quegli ormoni che mancano a causa della carenza di corticotropina (ACTH). Sono farmaci che vengono ingeriti per via orale due o tre volte al giorno.
Un farmaco chiamato levotiroxina viene utilizzato per sostituire una carenza di ormone stimolante la tiroide (TSH)..
Potrebbe esserci un deficit di ormoni sessuali. Per raggiungere livelli normali, agli uomini viene somministrato testosterone in modi diversi. Ad esempio, attraverso la pelle con un cerotto, con un gel per uso quotidiano o mediante iniezioni.
Nelle donne, estrogeni e progesterone vengono aggiunti al corpo con gel, cerotti o pillole. I contraccettivi orali sono i più utilizzati nelle giovani donne, mentre l'estradiolo valerato è consigliato per le donne prossime alla menopausa.
Quando c'è un deficit nell'ormone della crescita, è necessario iniettare la somatropina sotto la pelle. Coloro che ricevono questo trattamento in età adulta noteranno evidenti miglioramenti, sebbene non aumenteranno la loro altezza..
D'altra parte, se ci sono problemi di fertilità causati dal panipopituitarismo, l'iniezione di gonadotropine è possibile per stimolare l'ovulazione nelle donne. Così come la generazione di sperma negli uomini.
La stretta aderenza al trattamento è importante per migliorare. Come un follow-up nel tempo da parte di uno specialista endocrino. Ciò verificherà che il trattamento sia efficace e che i livelli ormonali rimangano nella normalità..
Nei casi in cui sono presenti tumori che hanno prodotto panipopituitarismo, è necessario un intervento chirurgico per rimuoverli. Se invece la ghiandola pituitaria è sotto pressione, si può scegliere una decompressione attraverso la chirurgia transfenoidale (bypassando l'osso sfenoidale). Quest'ultimo trattamento è il più indicato per il trattamento dell'apoplessia ipofisaria..
È stato dimostrato che una rapida decompressione potrebbe ripristinare parzialmente o completamente la funzione ipofisaria. Oltre a ridurre la necessità di terapia ormonale cronica (Onesti, Wisniewski & Post, 1990).
I pazienti con panipopituitarismo sembrano avere il doppio del rischio di morte. Principalmente a causa di affezioni respiratorie e cardiovascolari. Tuttavia, se viene rilevato precocemente e viene seguito il trattamento, il paziente può condurre una vita normale.



Nessun utente ha ancora commentato questo articolo.