Il facomatosi è un gruppo di malattie neurocutanee di origine genetica, rare nella popolazione generale. A livello clinico, sono caratterizzati dallo sviluppo di un coinvolgimento organico multisistemico con lesioni cutanee o tumorali, in diverse aree della pelle, degli organi o del sistema nervoso..
Inoltre, il suo decorso clinico aspecifico rende difficile la sua diagnosi precoce, quindi le sue conseguenze mediche e psicologiche deteriorano significativamente la qualità della vita della persona colpita e dei suoi parenti..
Sebbene ci sia un gran numero di malattie neurocutanee, le più comuni includono la fibromatosi di tipo I e II, la malattia di Bourneville, la sindrome di Sturge-Weber e la malattia di Von Hippel-Lindau..
D'altra parte, nonostante tutte queste siano patologie congenite, sono stati progettati molteplici approcci terapeutici di natura dermatologica che cercano di migliorare i segni e sintomi caratteristici di questi disturbi e, quindi, la prognosi medica di chi ne è affetto..
Indice articolo
Il termine facomatosi deriva dall'espressione di origine greca Phakos il cui significato si riferisce a <
Le patologie neurocutanee sono fondamentalmente caratterizzate dall'esistenza di un'associazione significativa tra un'affettività o disturbo neurologico e le manifestazioni dermatologiche.
Pertanto, il termine patologia neurocutanea viene utilizzato in modo generalizzato per comprendere diverse malattie che sono presenti nella persona congenita e che, inoltre, possono essere presenti per tutta la vita con lo sviluppo di lesioni cutanee e tumori in diverse aree, sistema nervoso, sistema cardiovascolare, sistema renale, sistema cutaneo, sistema oftalmico, ecc..
In questo modo il termine facomatosi fu introdotto nel 1917 da Brouwer e successivamente da van der Hoeve nel 1923, tuttavia le descrizioni iniziali facevano riferimento solo ad alcune patologie comprese in questo gruppo. Attualmente, più di 40.
A livello clinico, la facomatosi è descritta come una malattia che si presenta con alterazioni cutanee e malformazioni benigne / maligne in diversi sistemi: neurologico, oculare, cutaneo e viscerale..
Per quanto riguarda le zone colpite, vari autori sottolineano che quelle di origine ectodermica sono le più colpite, cioè la pelle e il sistema nervoso, sebbene possano interessare anche altri sistemi o dispositivi, come l'occhio.
Le sindromi e le patologie di origine neurocutanea sono malattie rare nella popolazione generale, sebbene non ci siano dati specifici a livello generale su tutte queste.
Pertanto, l'epidemiologia di questi disturbi varia a seconda del tipo di malattia, in particolare, la neurofibromatosi è una delle più comuni, con una prevalenza relativa di un caso ogni 300.000 nati.
Le malattie neurocutanee sono caratterizzate dallo sviluppo di lesioni cutanee. Nello specifico, la facomatosi si distingue da molte altre per la presenza di amartomi..
Gli amartomi sono un tipo di malformazione o tumore benigno che può crescere in diversi organi come il cervello, il cuore, gli occhi, la pelle o i polmoni..
Tuttavia, la facomatosi può essere associata a un ampio numero di condizioni mediche che variano, fondamentalmente, a seconda della specifica malattia o patologia sofferta dalla persona colpita..
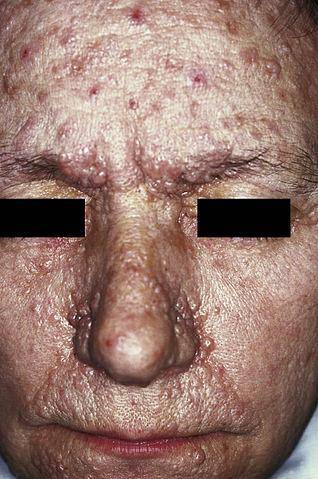
Attualmente, un gran numero di disturbi neurocutanei sono stati identificati a livello clinico e genetico, tuttavia ce ne sono alcuni con una maggiore prevalenza nella popolazione generale: neurofibromatosi di tipo I e tipo II, malattia di Bourneville, malattia di Von Hippel-Lindau e Sturge-Weber sindrome.
Esistono diverse forme cliniche di neurofibromatosi. Tuttavia, attualmente le più frequenti sono la neurofibromatosi di tipo I, chiamata anche malattia di Von Reclinghausen, e la neurofibromatosi di tipo II, seguita dalla shwannomatosi spinale..
A livello eziologico, tutte queste manifestazioni mediche della neurofibromatosi hanno un'origine genetica e si verificano con la formazione di tumori nelle aree nervose, in particolare nel sistema nervoso centrale e periferico..
Le formazioni tumorali, di solito non cancerose o benigne, tendono a crescere e svilupparsi quasi ovunque nel sistema nervoso, come il cervello, il midollo spinale o i nervi periferici.
Pertanto, le alghe delle complicanze mediche secondarie della neurofibromatosi includono anomalie nella crescita, sviluppo di convulsioni, comparsa di tumori cerebrali, patologie ossee, sordità e / o cecità o sviluppo di significativi problemi di apprendimento, tra gli altri.
Inoltre, questa patologia è presente dal momento della nascita. Tuttavia, la manifestazione significativa del suo quadro clinico può essere ritardata fino alla tarda infanzia, alla prima adolescenza o all'età adulta..
D'altra parte, la diagnosi di questo tipo di patologia comprende solitamente, oltre all'esame fisico e neurologico, diversi test di neuroimaging e analisi genetiche..
Inoltre, attualmente non esiste una cura per la neurofibromatosi, tuttavia, esistono approcci terapeutici specializzati nel controllo dell'affettività dermatologica, possono includere trattamenti sia farmacologici che chirurgici per fermare o eliminare le formazioni tumorali.
La neurofibromatosi di tipo I (NF1), nota anche come malattia di von Recklinghausen, si manifesta principalmente attraverso la presenza di macchie marrone chiaro, comunemente chiamate "café au lait", efelidi (lentiggini) e neurofibromi (danni ai nervi nelle cellule di Schwann e nei neuriti).
Ha un'origine genetica autosomica dominante, in particolare a causa di una mutazione sul cromosoma 17, nella posizione 17q11.2. Pertanto, il gene coinvolto in
lo sviluppo della neurofibromatosi di tipo I, ha un ruolo di primo piano nella modulazione della crescita e del differenziamento cellulare e, inoltre, può funzionare come soppressore del tumore.
Per quanto riguarda l'epidemiologia di questa patologia, presenta una prevalenza approssimativa di un caso ogni 2.500.3000 nati.
La diagnosi di neurofibromatosi di tipo I viene solitamente effettuata sulla base dei criteri clinici di consenso del National Institute of Health (1987), tuttavia richiede un follow-up continuo per evitare complicazioni mediche secondarie..
Normalmente, le escrescenze tumorali vengono trattate con farmaci, per prevenirne lo sviluppo esponenziale o mediante rimozione chirurgica.
La neurofibromatosi di tipo II (NF2), si manifesta principalmente attraverso lo sviluppo di schwannomi, cioè formazioni tumorali derivate dalle cellule di Shcwaan che saranno responsabili della copertura dei prolungamenti nervosi.
Schwannomi o neuriomi di solito colpiscono soprattutto il nervo uditivo e ottico e in misura minore le aree cutanee.
La neurofibromatosi di tipo II ha un'origine genetica autosomica dominante, in particolare a causa della presenza di una mutazione sul cromosoma 22, nella posizione 22q11.22.
Il gene coinvolto nello sviluppo di questa patologia è responsabile della codifica di una componente proteica con un ruolo di primo piano nella soppressione del tumore, quindi la sua attività carente produce un aumento anormale della proliferazione cellulare.
Per quanto riguarda l'epidemiologia di questa patologia, è meno frequente del tipo 1, presentando una prevalenza approssimativa di un caso ogni 50.000 nati.
La diagnosi di neurofibromatosi di tipo II è simile al tipo precedente e di solito viene effettuata sulla base dei criteri clinici di consenso dell'Istituto Superiore di Sanità. Tuttavia, di solito include test di laboratorio complementari, come il neuroimaging..
Normalmente, le escrescenze tumorali vengono trattate con farmaci, tuttavia, nei casi in cui è possibile, viene utilizzata la rimozione chirurgica.
La malattia di Bourneville è uno dei termini usati per riferirsi alla sclerosi tuberosa, una malattia genetica caratterizzata dalla presenza di amartomi.
A livello clinico può dar luogo ad una affettazione multisistemica caratterizzata da affettività cutanea (angiomi facciali, fibromi ungueali, placche fibrose, macchie ipocromiche, ecc.), Affezione renale (angiomiolipomi renali o cisti renali), affezione cardiaca (rabdomiomi cardiaci) , affettività neurologica (tuberi corticali, noduli gliali subependimali, atrocitomi, convulsioni, disabilità intellettiva, anomalie comportamentali e motorie), tra gli altri.
Come le malattie sopra descritte, l'origine della sclerosi tuberosa è genetica. In particolare, è dovuto alla presenza di mutazioni nei geni TSC1 e TSC2..
D'altra parte, la diagnosi di sclerosi tuberosa viene effettuata sulla base dei criteri clinici proposti in una conferenza medica nel 1998. Tuttavia, lo studio genetico è considerato rilevante anche per la sua conferma.
Per quanto riguarda il trattamento della sclerosi tuberosa, sebbene non esista una cura, vengono solitamente utilizzati diversi approcci farmacologici e chirurgici, principalmente per il controllo della crescita tumorale e delle complicanze mediche secondarie come le manifestazioni neurologiche.
La malattia di Von Hippel-Lindau, nota anche come angiomatosi retino-cerebellare, si manifesta principalmente attraverso la presenza e lo sviluppo di malformazioni vascolari, cisti e / o tumori, generalmente benigni..
Ha un'origine genetica autosomica dominante, in particolare a causa di una mutazione sul cromosoma 3, nella posizione 3p-25-26. Inoltre, presenta un'incidenza stimata di un caso ogni 40.000 nati..
Nello specifico, la malattia di Von Hippel-Lindau colpisce principalmente il sistema nervoso centrale (SNC) e la retina, attraverso la formazione di emangiomi.
Gli emangiomi sono malformazioni vascolari caratterizzate dalla presenza di grappoli di capillari sanguigni dilatati. Di solito compaiono nel cervello e nelle aree spinali, sebbene siano frequenti anche nelle retine o sulla pelle.
La diagnosi di questa patologia, oltre all'esame fisico e neurologico, richiede uno studio oftalmologico dettagliato, unitamente all'analisi di diversi test di neuroimaging, per confermare la presenza di lesioni nervose..
Per quanto riguarda il trattamento della malattia di Von Hippel-Lindau, l'intervento di base è un intervento chirurgico per eliminare le malformazioni vascolari. Tuttavia, richiede un monitoraggio continuo per evitare complicazioni secondarie..
Inoltre, ha un'aspettativa di vita ridotta, intorno ai 50 anni, principalmente a causa dello sviluppo di carcinomi a cellule renali (formazioni neoplastiche di cellule tumorali nei tubuli renali).
La sindrome di Sturge-Weber, nota anche come angiomatosi encefalotigeminale, si manifesta principalmente attraverso la presenza di emangiomi.
Un emangioma è un tipo di neoplasia o formazione di tumori caratterizzata dalla presenza di un numero anormalmente elevato di vasi sanguigni nella pelle o in altri organi interni..
In particolare, a livello clinico, la sindrome di Sturge-Weber è caratterizzata dallo sviluppo di emangiomi facciali, emangiomi intracranici ed emangiomi coridici, congiuntivali, episcerali e glaucomatosi..
Ha un'origine genetica, in particolare a causa di una mutazione sul cromosoma 9, nella posizione 9q21, nel gene GNQ. Questa componente genetica ha un ruolo di primo piano nel controllo di fattori di crescita, peptidi vasoattivi e neurotrasmettitori (Orhphanet, 2014).
La diagnosi della sindrome di Sturge-Weber viene effettuata sulla base del sospetto clinico e di diversi test di laboratorio, come la tomografia computerizzata o la risonanza magnetica per immagini..
D'altra parte, in termini di trattamento, la terapia laser è in grado di ridurre la progressione di questa patologia e, inoltre, in molti casi eliminare completamente gli emangiomi..


Nessun utente ha ancora commentato questo articolo.